Chordom: Ein seltener Knochenkrebs der Schädelbasis und der Wirbelsäule
Chordome sind niedriggradige Krebserkrankungen, die überall an der Wirbelsäule und der Schädelbasis auftreten können. Es wird angenommen, dass sie aus dem Notochord entstehen, einer rudimentären Struktur, die für die Entwicklung der Wirbelsäule wichtig ist. Diese Tumore sind recht selten und treten jährlich bei etwa 1/1.000.000 Menschen auf. Von allen Krebstumoren, die vom Knochen ausgehen, machen sie weniger als 4 % aus. Chordome treten typischerweise im Alter zwischen 40 und 70 Jahren auf, wobei Männer gegenüber Frauen bevorzugt werden. Diese Tumore metastasieren selten im Körper, sind aber lokal invasiv und aggressiv.
Bei etwa einem Drittel der Patienten haben Chordome ihren Ursprung in der Schädelbasis. Kopfschmerzen, Doppeltsehen, abnorme Augenbewegungen, Schluckbeschwerden und Harninkontinenz sind häufige Symptome für Schädelbasis-Chordome. Obwohl der Versuch, Wirbelsäulenchordome in einem Stück zu entfernen (En-bloc-Resektion), der anerkannte Ansatz ist, können die meisten Schädelbasischordome mit dieser Methode nicht entfernt werden. Die Behandlung dieser Tumoren umfasst daher eine minimalinvasive chirurgische Resektion sowie eine Strahlentherapie. Diese Kombination führt bei vielen Patienten zu einer Remission, bei einigen ist jedoch noch eine weitere Behandlung erforderlich. Zu den Zielen der Behandlung gehören die Tumorkontrolle und die Erhaltung der Lebensqualität.
Die Neurochirurgen Daniel Kelly, MD, und Garni Barkhoudarian, MD, vom Pacific Neuroscience Institute in Santa Monica und Torrance, Kalifornien, sind anerkannte Experten für die minimalinvasive chirurgische Entfernung und Behandlung von Chordomen.
Pathogenese
Obwohl die zugrunde liegenden Ursachen des Chordoms weniger bekannt sind, haben Chordome ihren Ursprung im Knochen, und während sie wachsen, zerstören sie den Knochen und breiten sich auf das Gehirn und das Weichgewebe aus. Der primäre Entstehungsort ist der Clivus (daher „clivale Chordome“), der eine Verbindung zwischen zwei Schädelknochen, dem Keilbein und dem Hinterhauptbein, darstellt. Viele Tumore durchbrechen die Dura mater – die Hülle von Gehirn und Rückenmark – und können direkten Druck auf den Hirnstamm und die Hirnnerven ausüben. Ebenso können die Tumore in die Nasennebenhöhlen einwachsen und Verstopfungen verursachen, die zu Sinusitis, Infektionen, Blutungen oder Funktionsstörungen der Eustachischen Röhre führen können.
Symptome
Die klinischen Erscheinungsformen von Chordomen unterscheiden sich je nach Lage und Größe des Tumors. Zu den häufigsten Symptomen von Schädelbasis-Chordomen gehören Kopfschmerzen (ähnlich wie Sinuskopfschmerzen), Nasennebenhöhlenentzündungen, Nasenbluten (Epistaxis) und Hörstörungen aufgrund einer Blockade oder Funktionsstörung der Eustachischen Röhre. Wenn der Tumor in das zentrale Nervensystem (ZNS) eindringt, kann er Symptome einer Hirnstammkompression und Nervenfunktionsstörungen verursachen. Der am häufigsten betroffene Nerv ist der Nervus abducens, der die Augenbewegungen steuert und zu Doppelbildern (Diplopie) führt. Weitere Nerven, die betroffen sein können, sind der Nervus trigeminus (Gesichtsempfindung), der Nervus facialis (Gesichtsmuskelbewegung), der Nervus vestibulocochlearis (Gehör und Gleichgewicht), der Nervus opticus (Sehstörungen) und der Nervus hypoglossus (Zungenbewegung). Bei schwerer Kompression des Hirnstamms können die Patienten Schluckbeschwerden (Dysphagie), eine heisere Stimme (Stimmbandstörung), Schwäche in Armen oder Beinen sowie Gleichgewichts- oder Koordinationsstörungen entwickeln.
Die Hypophyse befindet sich im oberen Teil des Clivus und kann manchmal durch den Tumor komprimiert oder verdrängt werden. Die Hypophyse ist die Hauptdrüse des Körpers und steuert andere endokrine Drüsen wie die Nebenniere und die Schilddrüse. Daher kann eine Hypophysenfehlfunktion zu Symptomen einer Hypothyreose oder eines Hypokortisolismus mit Symptomen wie Müdigkeit, Gewichtszunahme, Ausbleiben des Menstruationszyklus und Impotenz/Unfruchtbarkeit führen.
Bei einigen Patienten wird aufgrund von Symptomen, die nichts mit der Hypophyse zu tun haben, wie z. B. einem Kopftrauma, eine Schädelaufnahme durchgeführt, und es kann eine Läsion im Clivus festgestellt werden. Diese Tumore können asymptomatisch sein und wären sonst nicht entdeckt worden. Angesichts der Möglichkeit des Tumorwachstums und der Entwicklung neuer Symptome sollten sie gründlich untersucht und möglicherweise behandelt werden.
Diagnose
Chordome werden zunächst durch bildgebende Verfahren des Schädels diagnostiziert, am häufigsten durch Computertomographie (CT) und Magnetresonanztomographie (MRT). Häufig haben Patienten Symptome im Bereich der Nasennebenhöhlen und unterziehen sich einer CT des Kopfes durch ihren HNO-Arzt, was weitere Untersuchungen nach sich zieht. Der Goldstandard der Bildgebung ist eine MRT mit intravenösem Kontrastmittel, das Gadolinium verwendet, mit dünnen Schnitten im Bereich der Hirnanhangsdrüse, die oft als „Hypophysenprotokoll“ bezeichnet wird.
Wenn die Bildgebung zeigt, dass der Tumor die Hypophyse oder das Chiasma opticum zusammendrückt, können weitere Untersuchungen erforderlich sein, einschließlich einer endokrinologischen und/oder ophthalmologischen Beurteilung. Die Hormone der Hypophyse sollten bestimmt werden und erfordern möglicherweise eine gründlichere Untersuchung. Ein Gesichtsfeldtest und eine optische Kohärenztomographie (OCT) sollten in Betracht gezogen werden, um das Ausmaß des Sehkraftverlusts und der dauerhaften Schädigung des Sehnervs zu messen.
Da es zahlreiche Arten von Tumoren gibt, die in dieser Region auftreten können, mit sehr unterschiedlichen Behandlungsmöglichkeiten, ist fast immer eine erste Biopsie erforderlich, um eine pathologische Diagnose zu stellen. Diese wird häufig vom HNO-Chirurgen im Rahmen eines ambulanten oder stationären Eingriffs durchgeführt. Die pathologische Diagnose eines Chordoms ist in der Regel eindeutig und notwendig, um die endgültige Behandlung des Patienten festzulegen.
Behandlung von Chordomen am PNI
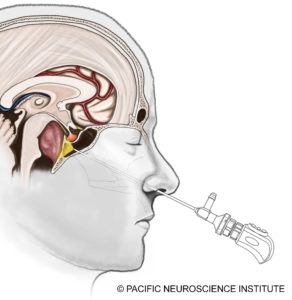 Am Pacific Neuroscience Institute und anderen Top-Zentren ist der Goldstandard für die Behandlung von Chordomen die chirurgische Resektion des Tumors. Die meisten Chordome an der Schädelbasis liegen in der Mittellinie und lassen sich am besten mit einem minimalinvasiven Ansatz wie einem endoskopischen endonasalen transklivalen Zugang durch die Nasenlöcher behandeln. Einige Tumore haben eine seitliche Invasion, z. B. in die Innenohrregion, und können eine retrosigmoidale Kraniotomie erfordern, bei der der Tumor in Kombination oder allein von hinter dem Ohr aus behandelt wird. Das Ziel der Operation ist die maximale sichere Resektion des Tumors. Eine vollständige Tumorresektion sollte nicht angestrebt werden, wenn lebenswichtige Funktionen gefährdet sind.
Am Pacific Neuroscience Institute und anderen Top-Zentren ist der Goldstandard für die Behandlung von Chordomen die chirurgische Resektion des Tumors. Die meisten Chordome an der Schädelbasis liegen in der Mittellinie und lassen sich am besten mit einem minimalinvasiven Ansatz wie einem endoskopischen endonasalen transklivalen Zugang durch die Nasenlöcher behandeln. Einige Tumore haben eine seitliche Invasion, z. B. in die Innenohrregion, und können eine retrosigmoidale Kraniotomie erfordern, bei der der Tumor in Kombination oder allein von hinter dem Ohr aus behandelt wird. Das Ziel der Operation ist die maximale sichere Resektion des Tumors. Eine vollständige Tumorresektion sollte nicht angestrebt werden, wenn lebenswichtige Funktionen gefährdet sind.
Die endonasale Operation durch die Nase ist heute der Hauptpfeiler der chirurgischen Möglichkeiten und ist in den letzten 10 Jahren immer sicherer geworden. Mit dem Einsatz von chirurgischer Navigationssoftware, d.h. „GPS“ für die Operation, Doppler-Ultraschallüberwachung und Hirnnervenüberwachung sind die Risiken von Gefäßverletzungen, einschließlich Blutungen oder Schlaganfall, und Nervenverletzungen zurückgegangen. Darüber hinaus hat die Einführung eines „nasoseptalen“ vaskularisierten Lappens, bei dem normales Sinusgewebe zur Abdeckung des chirurgischen Defekts bewegt wird, die Rate der Liquorlecks nach der Operation erheblich verringert. Postoperative Liquorlecks können, wenn sie nicht behandelt werden, zu einer Meningitis führen, die tödlich sein kann. In den letzten sieben Jahren hatten wir keinen Liquoraustritt nach einer Chordom-Operation.
 Wenn der Tumor durch die Operation vollständig entfernt wurde, ist keine weitere Therapie erforderlich, es sei denn, der Tumor wächst erneut. Im Falle eines Resttumors, d. h. eines Tumors, der nicht sicher entfernt werden konnte, kann jedoch eine zusätzliche Behandlung erforderlich sein, insbesondere wenn es Anzeichen für ein Wachstum gibt. Zu den besten Behandlungsmöglichkeiten gehören die stereotaktische Radiochirurgie oder die Protonenbestrahlung oder eine Kombination aus beidem. Diese Optionen werden in Zusammenarbeit mit unseren Kollegen aus der Strahlenonkologie durchgeführt. Dem Radiochirurgen kommt die wichtige Aufgabe zu, den zu behandelnden Tumor zu identifizieren und gefährdete Strukturen nach Möglichkeit zu schützen. Sowohl die stereotaktische Radiochirurgie als auch die Protonenbestrahlung bieten eine angemessene Tumorkontrolle von etwa 80 Prozent, wobei die Protonenbestrahlung je nach Lage des Tumors leicht im Vorteil ist.
Wenn der Tumor durch die Operation vollständig entfernt wurde, ist keine weitere Therapie erforderlich, es sei denn, der Tumor wächst erneut. Im Falle eines Resttumors, d. h. eines Tumors, der nicht sicher entfernt werden konnte, kann jedoch eine zusätzliche Behandlung erforderlich sein, insbesondere wenn es Anzeichen für ein Wachstum gibt. Zu den besten Behandlungsmöglichkeiten gehören die stereotaktische Radiochirurgie oder die Protonenbestrahlung oder eine Kombination aus beidem. Diese Optionen werden in Zusammenarbeit mit unseren Kollegen aus der Strahlenonkologie durchgeführt. Dem Radiochirurgen kommt die wichtige Aufgabe zu, den zu behandelnden Tumor zu identifizieren und gefährdete Strukturen nach Möglichkeit zu schützen. Sowohl die stereotaktische Radiochirurgie als auch die Protonenbestrahlung bieten eine angemessene Tumorkontrolle von etwa 80 Prozent, wobei die Protonenbestrahlung je nach Lage des Tumors leicht im Vorteil ist.
Bislang hat sich kein Medikament als „Wunderwaffe“ zur Behandlung von Chordomen erwiesen. Es gibt jedoch einige vielversprechende Optionen, vor allem wenn genetische Zielstrukturen identifiziert werden. Selbstverständlich werden die Chordome unserer Patienten einer gründlichen genetischen Sequenzierung unterzogen, um die Genmutationen zu analysieren. Wenn der Patient ein Kandidat für eine gezielte Therapie ist, kann dies eine Option für unsere Neuroonkologen sein. Am Saint John’s Cancer Institute führen wir eine Reihe von Studien für Chordome durch, in denen wir potenzielle Behandlungsmöglichkeiten für Chordom-Patienten untersuchen.
Schlussfolgerung
Chordome können mit subtilen Symptomen auftreten und ein schleichendes Wachstum aufweisen. Die Behandlungsmöglichkeiten sind die Tumorbiopsie, gefolgt von einer minimalinvasiven Tumorresektion. Bei einigen Patienten kann eine Strahlentherapie erforderlich sein, wenn der Tumor nach der Operation weiter wächst. Die Spitzenforschung hat mehrere potenzielle Medikamente identifiziert, die Patienten mit rezidivierenden Chordomen einen gewissen Nutzen bieten können. Mit unserem Team aus Neurochirurgen, HNO-Chirurgen, Augenärzten, Endokrinologen, Strahlentherapeuten und Neuroonkologen sind wir am PNI und am Saint John’s Cancer Institute in der Lage, Chordom-Patienten einen umfassenden Ansatz zu bieten.
Für weitere Informationen oder ein Beratungsgespräch mit einem unserer Experten erreichen Sie uns unter 310-582-7450.

Garni Barkhoudarian, MD, ist Neurochirurg mit Schwerpunkt Schädelbasis und minimal-invasive endoskopische Chirurgie. Sein besonderes Interesse und Fachwissen gilt Hypophysen- und Parasellartumoren, Hirntumoren, Schädelbasistumoren, intraventrikulären Hirntumoren, Kolloidzysten, Trigeminusneuralgie und anderen vaskulären Kompressionssyndromen. Er ist an einer Reihe von klinischen Studien beteiligt, in denen die Wirksamkeit verschiedener medikamentöser oder chemischer Therapien für Hypophysentumore und bösartige Hirntumore untersucht wird. Bei praktisch allen Tumoren und intrakraniellen Eingriffen wendet Dr. Barkhoudarian das Schlüsselloch-Konzept zur Minimierung von Kollateralschäden am Gehirn und seinen unterstützenden Strukturen an, indem er fortschrittliche Neurobildgebungs- und Neuronavigationstechniken zusammen mit der Endoskopie einsetzt, um die Zielgenauigkeit und die Visualisierung von Läsionen zu verbessern.